Developmental Neuropathology
Chapter 27.1: Disorders of Carbohydrate Metabolism: Lysosomal Disorders
Catabolic pathways: lysosomal storage disorders, e.g., mucopolysaccharidoses (MPS), oligosaccharidoses, type II glycogenosis
- single system disorders
- direct morphological manifestations
- easier to study
Anabolic pathways: congenital defects in glycosylation (CDG)
- multisystem disorders
- often nonspecific morphological features
- harder to study
Lysosomal disorders
1. Mucopolysaccharidoses (MPS)
2. Oligosaccharidoses
3. Mucolipidoses
4. Type II dlycogenosis
Defining features of lysosomal disorders:
- Lysosomes are vacuolated
- Lysosomal vacuolar contents are glycosaminoglycams
- Lysosomal storage in nearly every organ and cell type
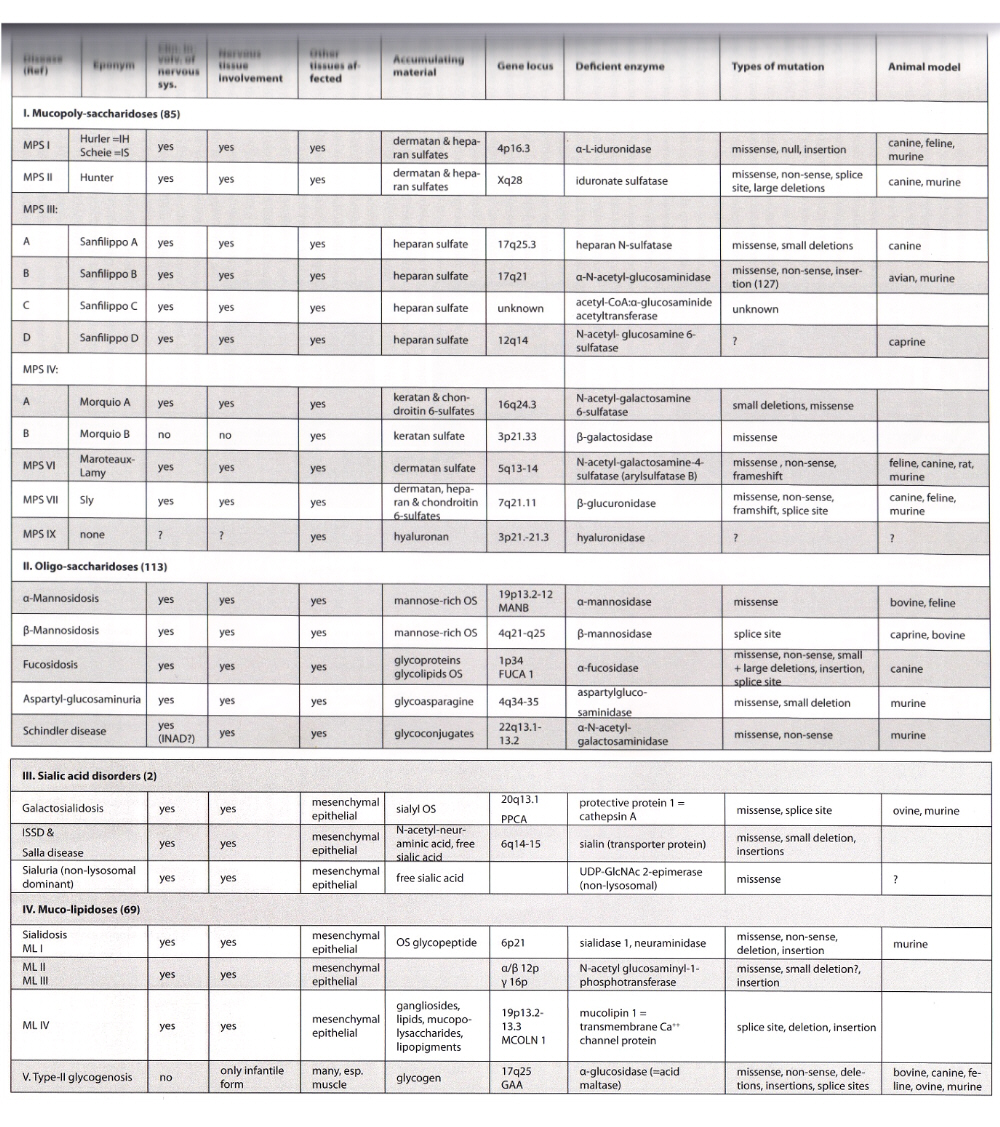
- MPS V has been eliminated as it has been found to be allelic to MPS I (Scheie type)
- MPS IV-B involves lysosomal beta-galactosidase, and is allelic to GM1-gangliosidosis
- mucolipidosis II also called I cell disease (for "inclusions" in cultured cells)
- mucolipidosis III also called pseudo-Hurler polydystrophy
- infantile type II glycogenosis is also called Pompe's disease
- epidemiology varies between disorders, but most are rare, with some geographical variation, e.g., Salla disease and aspartyl-glucosaminouria are more common in Finland
- all are autosomal recessive, except MPS II which has X-linked inheritance
Embryology
- lysosomal storage begins before birth
- mental retardation
- skeletal deformities - dysostosis multiplex
- organomegaly
- coarse face, short stature
- milder forms can be delayed
Clinical features
Mucopolysaccharidoses
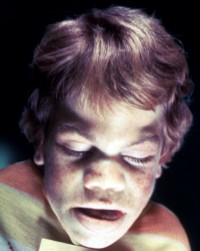
- MPS I (Hurler phenotype)
- normal at birth, early psychomotor retardation
- coarsening of facial features, skeletal changes, hepatospelnomegaly, frequent ear infections
- mental reardation and delcine, growth retardation, restriction of joint mobility, carpal tunnel syndrome
- corneal clouding, macrocephaly, hydrocephalus from meningeal thickening
- life expectancy <2 decades
- radiological findings of dysostosis multiplex
- MPS I (Scheie phenotype)
- milder, normal height and mentation
- mild skeletal changes
- carpal tunnel and cardiac valve disease
- MPS II (Hunter) - X-linked
- same as Hurler, except no corneal clouding
- MPS III
- mainly neurological features, with mental retardation and psychomotor regression and hyperactive behaviour
- thick hair, minimal skeletal changes
- MPS IV and V
- no direct CNS involvement, mostly skeletal
- can have hydrocephalus from meningeal thickening
- can have cord compression from atlanto-axial dislocation
- can have carpal tunnel sundrome
- MPS VII
- very rare, like Hurler, but spectrum from hydrops fetalis to almost unaffected
Oligosaccharidoses
- Mannosidosis
- features similar to Hurler, but variable severity
- hearing loss
- Fucosidosis
- wide picture
- severe neurological picture, mild skeletal changes, hepatomegaly
- angiokeratoma of the skin is suggestive but not specific
- Aspartylglycosaminuria
- mainly in Finland
- language delay, mental retardation
- mild facial coarsening amd skeletal abnormalities
- Galactosialidosis
- severe disease in infancy resembling GM1-gangliosidosis and sialidosis
- juvenile form starts with extrapyramidal symptoms and ataxia, followed by myoclonus, mental decline
- mild skeletal features
- Salla disease
- mental retardation, ataxia, progressive psychomotor decline
- ISSD (infantile sialic storage disease)
- more severe than Salla disease, although same mutation
Mucolipidoses
- Sialidosis (mucolipidosis I)
- severe disorder in early childhood, or a juvenile form
- action polymyoclonus, ataxia, seizures, gradual mental decline and visual loss
- macular cherry red spot
- no organomegaly
- Mucolipidosis II
- coarse facial features and dysostosis myltiplex
- Mucolipidosis III
- milder than II, though same mutation
- Mucolipidosis IV
- mental retardation and visual decline with corneal opacities
Macroscopy
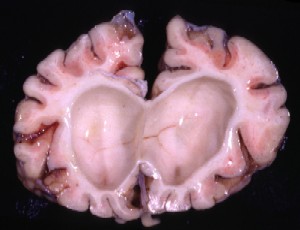
- thickening of the leptomeninges, sometimes occluding the outflow of CSF and hydrocephalus
- dura can also thicken, impairing the width of the spinal canal and compression
- wide Virchow-Robin spaces, especiallly in the white matter
- enlargement of the choroid plexus from lysosomal vacuolation
Histopathology
- wide-spread lysosomal vacuolation leading to intracellular vacuolation of mesenchymal cells
- cells affected include blood vessels
- zebra bodies - lamellar/membranous appearance of ganglioside-containing lysosomes
- late onset disease is associated with increased lipopigments in neurons (e.g., MPS III), including subunit C of mitochondrial ATP synthase (also seen in neuronal ceroid lipofuscinosis)
- neuronal storage especially in the subcortical grey matter, including the spinal cord
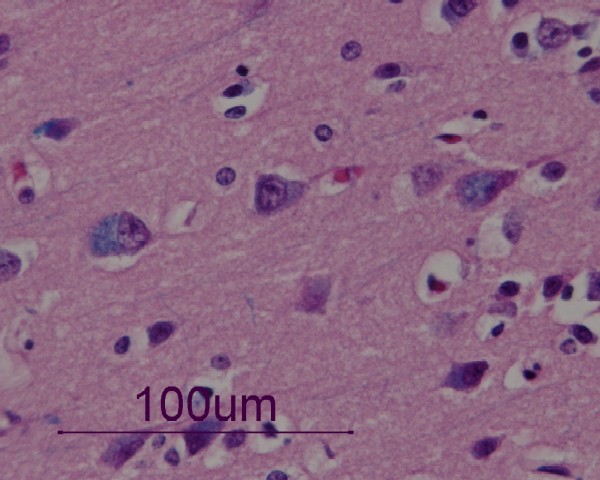
- meganeurite - can also affect the proximal axon
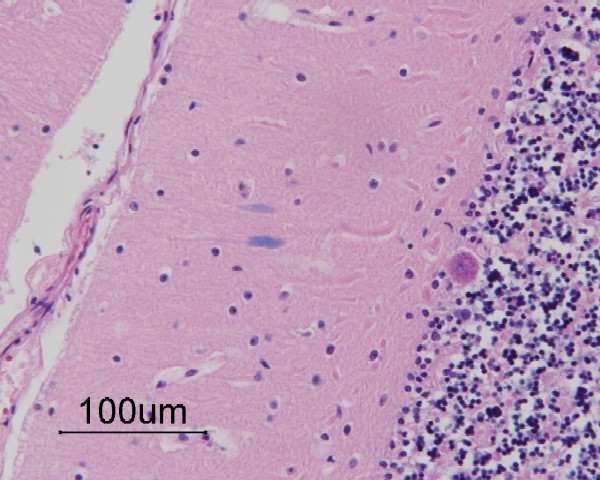
- asteroid body - lysosomal storage in dendrites of Purkinje cells
- neuronal loss and gliosis
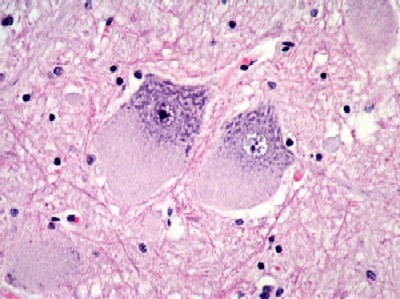
- type II glycogenosis
- ubiquitous storage of lysosomal glycogen in skeletal/heart muscles, nerve cells, glial cells, Schwann cells, etc.
- especially in subcortical grey matter and ballooned anterior horn cells of spinal cord
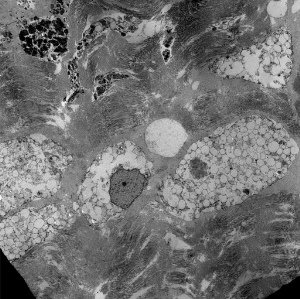
Immunohistochemistry and ultrastructural findings
- immunohistochemistry for lysosomal storage disorders not well developed
- ultrastructural findings useful in diagnosis
- lysosomal vacuoles with ill-defined contents
- may see glycogen granules
- "zebra bodies" in MPS
- lipopigments in the form of curvilinear profiles (like in NCL)
Lysosomal vacuoles and zebra bodies in mucopolysaccharidosis (mitral valve)
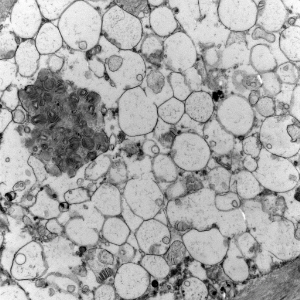
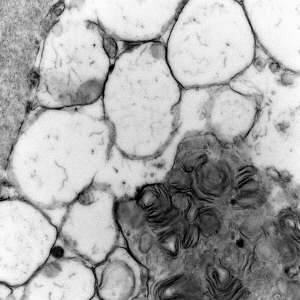
Lysosomal vacuoles in mucolipidosis (skin biopsy)
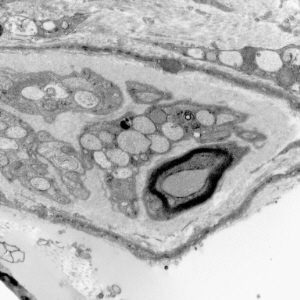
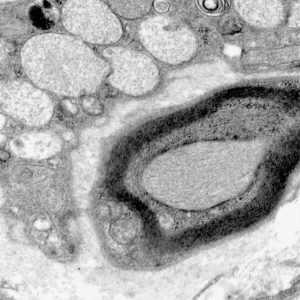
Biochemistry
- abnormal substrate levels from degradation disorders are excreted in the urine as mucopolysaccharides, oligosaccharides, and sialic compounds
- sometimes hydrolases can be measured in the serum or in cultured cells, e.g., fibroblasts
Differential Diagnosis
- ultrastructural analysis can divide between vacuolar and nonvacuolar lysosomal residual bodies
- defining the disorder requires clinical, biochemical, and molecular information
Pathogenesis
- reduced or absent lysosomal enzymes resulting in build up of substrates, damaging intracellular metabolism