Developmental Neuropathology
Chapter 27.2: Disorders of Carbohydrate Metabolism: Polyglucosan Disorders
Accumulation of polyglucosan bodies as intracellular extralysosomal carbohydrate-containing filament aggregates in various tissues in the body.
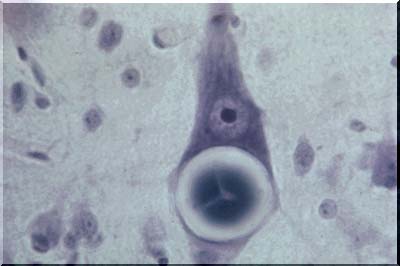
Lafora disease - Lafora bodies in nerve cells
Polyglucosan neuropathy - in axons
Polyglucosan body myopathy - in skeletal muscle
Polyglucosan body disease - in astrocytes
Type IV and VII glycogenosis - in astrocytes
Polyglucosan disorders can affect one organ or multiorgan systems.
Synonyms
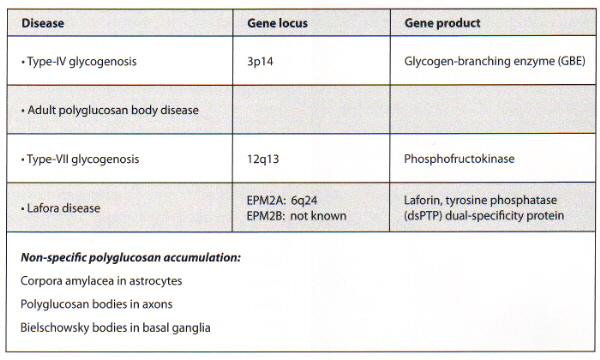
- Type IV glycogenosis - Andersen disease, amylopectinosis, brancher enzyme deficiency
- Type VII glycogenosis - Tarui disease, phosphofructokinase deficiency
- Lafora disease - myoclonus epilepsy (EPM2). EPM1 is myoclonus epilepsy of Unverricht-Lundborg without polyglucosan bodies
Epidemiology
- Adult polyglucosan body disease - Ashkenazi Jewish families with Y329S mytation in the glycogen-branching enzyme
- Lafora disease - in Mediterranean countries, and with founder effect, but also sporadic cases in Japan
- can be sporadic or autosomal recessive
- some manifest in infancy, others late in adult life
Genetics
Clinical Features
Adult polyglucosan body disease
- progressive dementia, upper and lower motor neuron defects, neurogenic bladder
- sensory deficits in both CNS/PNS
- spastic paraparesis and peripheral neuropathy
- rare frontal lobe involvement
- variants include a myopathic form and a severe infantile form (deficiency in branching enzyme and phosphorylase)
Type IV glycogenosis
- hepatosplenomegaly, liver cirrhosis, failure to thrive
Classical Lafora disease
- juvenile onset around 14 years
- epilepsy of many types including generalized tonic-clonic, occipital, absence-like seizures
- stimulus-sensitive polymyoclonus
- mental retardation and regression
- progressive with increasing myoclonus, ataxia, status epilepticus, loss of ambulation, mutism
- death by early twenties
Atypical Lafora disease
- a subset of patients with mutations in EPM2A
- initial learning difficulties with occasional seizures
- seizures becoming more common by 8-13 years of age, including myoclonic seizures, visual seizures, and status
- dementia, dysphagia, and respiratory problems later on
- this form likely more severe than classic Lafora disease
- another late onset atypical form exists
Macroscopy
- not usual to see gross findings
- in late-onset Lafora disease, can see wide-spread cortical necrosis or extensive calcification
- adult polyglucosan body disease can show brain atrophy and spinal cord atrophy, as well as demyelination
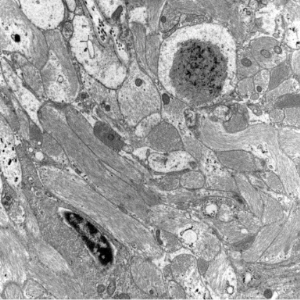
Histopathology
- polyglucosan bodies include a number of ultrastructurally similar inclusions
- can be nonspecific:
- in axons of the CNS and PNS related to aging
- corpora amylacea in astrocytic processes
- staining properties of polyglucosan bodies:
- stains with PAS, BEST's carmine for glycogen, iodine, Alcian blue
- not stained by Congo red
- can show metachromasia with toluidine blue
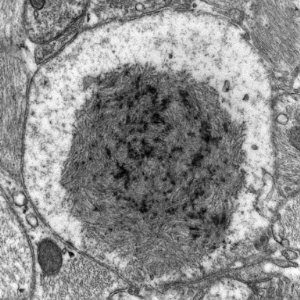
- Lafora bodies and corpora amylacea are spheroid
- Bielschowsky bodies are elongated, located in neuronal perikarya and dendrites, isolated to the basal ganglia, sometimes near status marmoratus (post perinatal hypoxic damage)
- infantile type IV glycogenosis
bodies in astrocytes in subpial, subependymal, and perivascular regions, both in grey and white matter
- Lafora disease
- Lafora bodies in grey matter in both neuronal perikarya and neuronal processes, mostly in the subcortical grey matter including brainstem and cerebellar nuclei
- loss of nerve cells corresponding to density of Lafora bodies
- can appear in ganglionic and bipolar cells of retina
- outside of CNS, in epithelial cells of eccrine sweat gland ducts, skeletal muscle fibers, and cells of the PNS
- Adult polyglucosan body disease
neuronal processes and astrocytes in both grey and white matter, also spinal cord and PNS
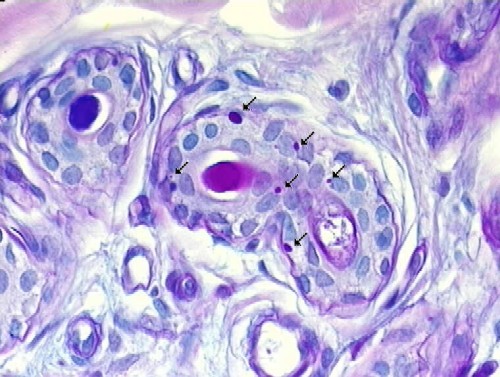
Lafora bodies seen in apocrine glands in a skin biopsy:
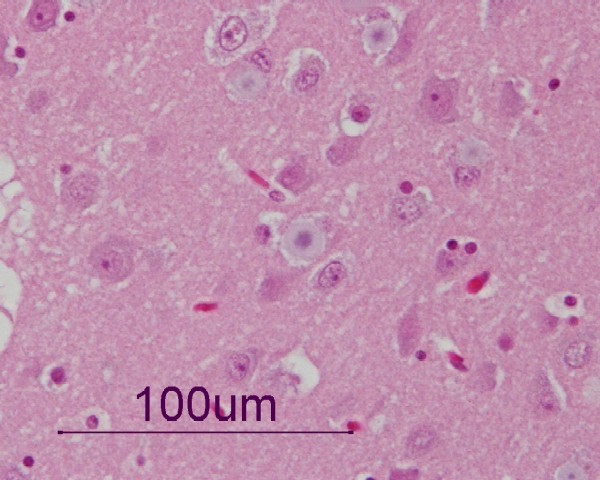
Lafora bodies in the cortex of a dog with myoclonic epilepsy:
Immunohistochemistry and Ultrastructural Findings
- Lafora body reacts with antibodies against ubiquitin
- KM-279 antibody also reacts with Lafora bodies, corpora amylacea, basophilic masses in the heart, filamentous glycogen inclusions in type IV glycogenosis, and Bielschowsky bodies
- 160kD and 200kD neurofilament, and desmin antibodies also react with Lafora bodies
- still unclear if mutant Laforin is in the Lafora bodies
- polyglucosan bodies - in axons
- Lafora bodies - in neuronal perikarya and dendrites
Lafora bodies (temporal lobectomy)
Biochemistry
- type IV glycogenosis - glycogen branching enzyme - cooperates with glycogen synthase to branch glycogen
- Lafora disease - laforin - dual-specificity protein tyrosine phosphatase (both phosphotyrosine and phosphoserine are dephosphorylated)
Pathogenesis
- Laforin binds glycogen at the N-terminus
- laforin may normally inhibit the action of glycogen synthase
- if so, mutant laforin would result in overactive glycogen synthase, producing a fibrillar glycogen that aggregates with proteins to form Lafora bodies
- still unclear how this negatively affects the cell, possibly by depleting energy sources
- similar mechanism may exist for glycogen branching enzyme deficiency