Developmental Neuropathology
Chapter 27.3: Disorders of Carbohydrate Metabolism: The Congenital Disorders of Glycosylation
Inborn errors of metabolism affecting the synthesis and processing of N-linked glycans. This includes all stages from synthesis of nucleotide-linked sugars to final processing in the Golgi apparatus. Technically, this also includes mucolipidoses II (I-cell disease) and III (pseudo-Hurler polydystrophy) due to affected synthesis of mannose-6-phosphate.
O-linked glycan synthesis defects also cause disease, e.g., progeroid form of Ehlers-Danlos sundrome, familial exostosis, Fukuyama muscular dystrophy, Muscle-Eye-Brain disease, Walker Warburg Syndrome, LGMD2I.
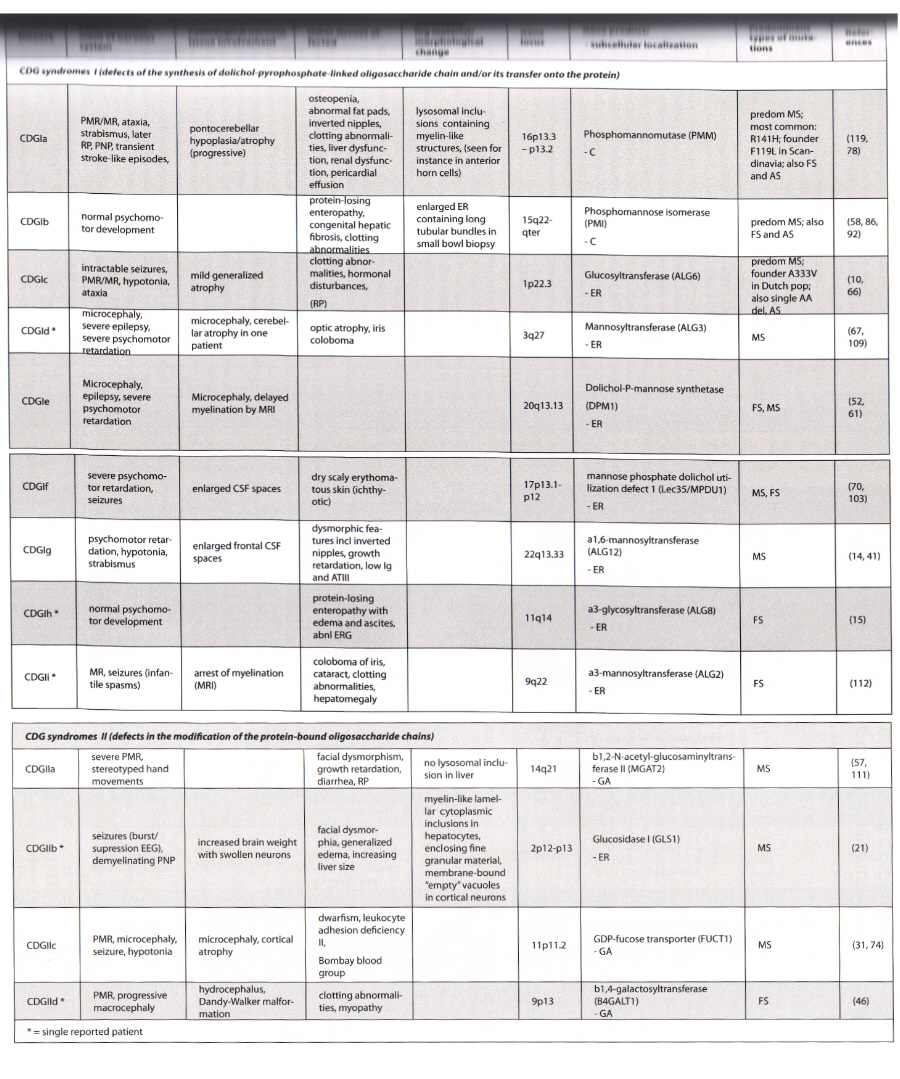
Congenital disorders of glycosylation are multisystem disorders, with prominent nervous system involvement. Type Ib has no CNS involvement.
Epidemiology
- CDG 1a is most common, with frequency of 1 in 40,000 to 60,000 in Sweden and Denmark
- incidence elsewhere unknown
- CDG 1c next most common
- other than 1b, other forms so rare that is likely underdiagnosed due to nonspecific symptoms
- all are autosomal recessive
- present congenitally or early childhood
- if survives early critical multisystem phase, can have more stable nonprogressive course into adulthood
Clinical Features
- Type Ia (most common and well known)
- present in early infancy with multisystem disease, hypotonia, alternating strabismus, feeding difficulties, hepatopathy, tubulopathy, pericardial effusions
- dysmorphism with inverted niples, unusually distributed subcutaneous fatpads on buttocks, joint contractures
- severity is variable
- initial mortality 20%
- if survive, psychomotor retardation, hypotnoia, ataxia, polyneuropathy, retinitis pigmentosa, transient stroke-like episodes, seizures
- osteopenia, skeletal deformities
- no evidence for degenerative process - stable after initial phase
- milder forms with borderline cognitive impairment
- neuroimaging shows pontocerebellar atrophy
- blood tests show coagulopathy with decreased AT III and factor XI, other endocrine abnormalities
- Type Ic - milder neurological involvement without cerebellar atrophy and little multisystem disease
- Type Ib - no major neurological involvement, protein losing enteropathy, hepatic fibrosis, hypoglycemia
Macroscopy
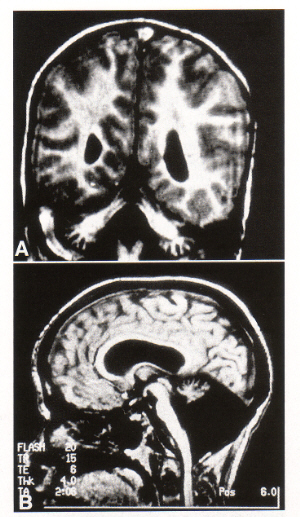
- type Ia shows shrunken cerebellum with firm, prominent foliae
- flattened pons
- cerebral hemispheres unremarkable
- general autopsy shows enlarged liver and kidneys
Histopathology
- gliotic cerebellar cortex with prominent loss of Purkinje and granular cells
- rare club-shaped expansions of Purkinje dendrites
- relative preservation of the dentate nucleus
- neuronal loss and gliosis in the pontine nuclei and transverse pontine fiber atrophy
- neuronal loss in the inferior olivary nucleus
- moderate cell loss in the cerebral cortex
- liver cirrhosis, fibrosis, fatty infiltration, giant cell hepatitis
Immunohistochemistry and Ultrastructural Findings
- axonal swellings with mitochondria and lamellar bodies have been observed in the ventral-medial and ventral-lateral thalamus
- myelin-like fibrillary lysosomal inclusions in hepatocytes, Schwann cells, and endoneural/epineural fibroblasts
- only defects in the ER or pre-ER stages result in lysosomal inclusions (not Golgi apparatus-based defects)
Differential Diagnosis
- other olivo-ponto-cerebellar atrophy (OPCA) conditions, including Norman's disease (granular layer aplasia) - may be allelic condition to CDG I
Biochemistry
- all the disorders affect N-glycan synthesis
- Types Ia and Ib both affect cytoplasmic synthesis of guanosine diphosphate mannose, needed for this glycosylation
- many glycoproteins affected, e.g., transferrin, which is used for diagnosis of CDG (not sensitive for type IIb and IIc, false positives with chronic alcohol consumption, galactosemia, and hereditary fructose intolerance)
- other proteins include alpha-1-antitrypsin and hexosaminidase
Pathogenesis
- glycosylation defects affect many secreted, membrane-bound, and intracellular proteins causing a wide range of symptoms
- N-linked glycosylation is important in protein stabilty, protein trafficking, protein function, molecular recognition, cell adhesion, and antigenicity
- complete interruption of the synthesis is incompatible with development
- high metabolic rate organs more affected with decreased enzymatic activity, e.g., liver
- unclear why CNS is so affected
Treatment
- Type Ib has been successfully treated with high dose mannose therapy
- this has not worked in type Ia
- Oral fucose therapy sucessful to treat CDG IIc