Developmental Neuropathology
Chapter 4&5: Lissencephaly
Lissencephaly: class of human cerebral malformations characterized by the agyric surface of the brain. Often the most ventral and medial gyri are relatively spared, as well as some other variable brain regions.
- underlying defect of cell migration - lissencephaly and heterotopia are clearly pathogenetically proven to be such
- slow/delayed cell migration
- results in a thickening of the cerebral cortex
- other associated malformations also seen
- lissencephaly, agyria, pacchygyria - spectrum of disorders - overlapping genetic mutations
- pachygyria:patchy or focal malformations
- agyria/lissencephaly: diffuse malformation of the cerebral hemispheres
Type I Lissencephaly: "classical lissencephaly"; lissencephalies associated with a failure of normal cell migration.
Type II Lissencephaly: "cobblestone lissencephaly"; lissencephalies associated with apparent over migration of neurons.
Type I Lissencephaly
- Incidence estimated at 1:500,000 (maybe underestimate)
- Equal M:F except for at least 2 X-linked forms
- No known risk factors, all known cases are genetic in origin
- Unclear if pachygyria may be due to environmental, toxic, or infectious etiology
Embryology
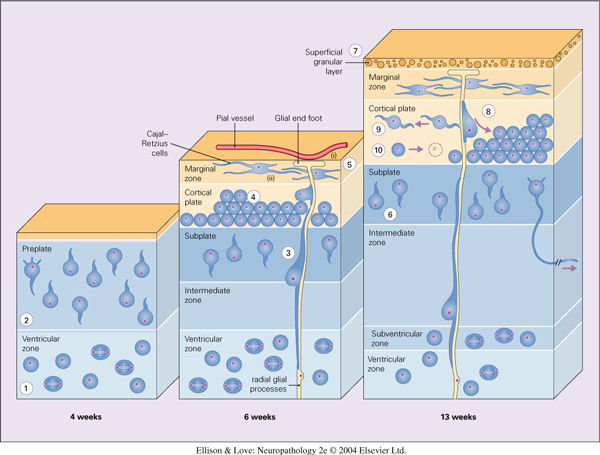
- defect in radial cell migration
- neuroblasts exit cell cycle and migrate outwards towards the brain surface
- neuroblasts are associated with radial glial cells
- radial glial cells are bipolar cells with a short process extending to the ventricle and a long process to the pial surface, forming a scaffold for radial cell migration. They are also neural stem cells.
- two way signaling between the neuroblasts and the radial glial cells to direct migration and maintain structure of the radial glial cells
- receptors involved, e.g., neuregulin, ErbB4, cell adhesion molecules, and their receptors
- blocking of any part of this can cause type I lissencephaly
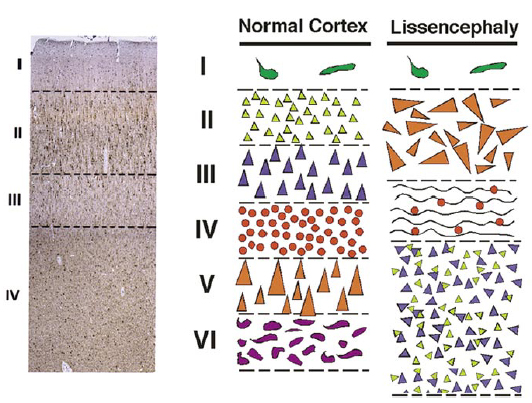
- mature cortex has 6 layers
- layers generated from inside to outside sequence
- first cells exit the ventricular zone (VZ) towards the cortical plate (future cortex) to form the pre-plate (outer Cajal-Retzius neurons, inner subplate neurons)
- subsequent cells from the ventricular zone enter between these two layers of the pre-plate in an inside to outside sequence (layers 6 to 1)
- importance of cells knowing when to stop migrating
- studied first in mice models, i.e., the Reeler mouse, where a mutation in the Reelin gene (expressed by Cajal-Retzius cells) causes inversion of the cortical layers
- human equivalent RELN found in some type I lissencephaly and cerebellar hypoplasia
Genetics
- Type I lissencephaly can be sporadic, autosomal dominant, autosomal recessive, or X-linked
- Four genes have been identified to date
- LIS1 (chr 17p) deletions in Miller-Dieker syndrome (MDS) or isolated lissencephaly sequence
- XLIS (chr Xq22.3-23) encoding for doublecortin (microtubule-associated protein) causes lissencephaly in males and subcortical band heterotopia in females (result of Lyonization)
- RELN is required for normal migration, mutations causing an autosomal recessive lissencephaly with cerebellar hypoplasia, with associated lymphedema and neuromuscular problems
- ARX is a transcription factor on the X chromosome that causes lissencephaly and ambiguous genitalia in males. Females have other symptoms but no lissencephaly
Clinical features
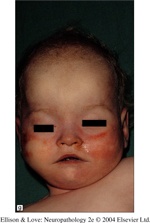
- overlapping features of all type I lissencephalies
- moderate to severe developmental delay and neuromotor impairment
- profound mental retardation and intractible seizures
- infantile spasms common in mutations in ARX
- isolated lissencephaly less severe than Miller-Dieker syndrome
- other associated features:
- acquired microcephaly
- mild dysmorphic facies
- MDS patients have bitemporal hollowing, hypertelorism, frontal bossing, short-upturned nose, small jaw, prominent upper lip with thin vermillion border, clinodactyly, polydactyly, cryptorchidism, sacral dimples, congenital heart defects
- ARX mutation results in microphalus, small adrenal glands - X-linked lissencephaly with ambiguous genitalia (XLAG) syndrome
- RELN mutation associated with congenital lymphedema
- imaging findings, particularly MRI
- loss of sulci and gyri, with thickened cerebral cortex
- heterotopia, subcortical bands, hydrocephalus, posterior fossa abnormalities
- pattern aids in guiding diagnosis
- LIS1 mutation shows relative sparing of anterior cortex
- XLIS mutation shows relative sparing of posterior cortex
- RELN mutation shows severe cerebellar hypoplasia
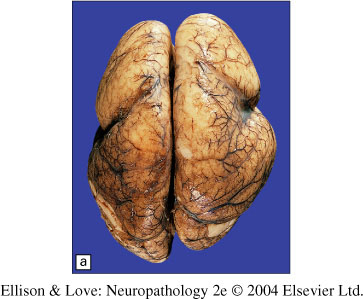
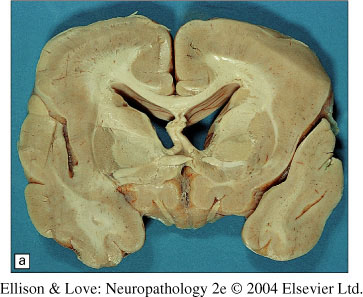
Macroscopic findings
- marked paucity of sulci/gyri, not necessarily generalized
- thickening of the cerebral cortex
- loss of underlying white matter
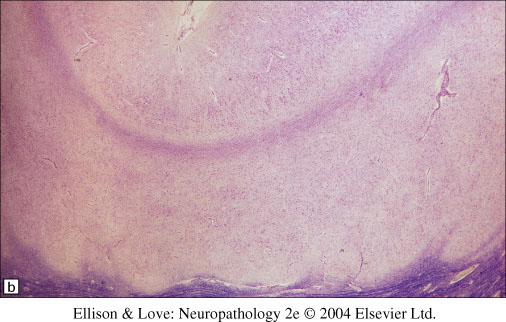
- transition areas with subcortical bands and heterotopia (periventricular and white matter)
- white matter band at layer 3
- large ventricles
- cerebellum may be hypoplastic
- inferior olivary dysplasia in LIS1 mutation
- multifocal pachygyria (thickened areas of cortex) can resemble lissencephaly, except it is not usually symmetrical
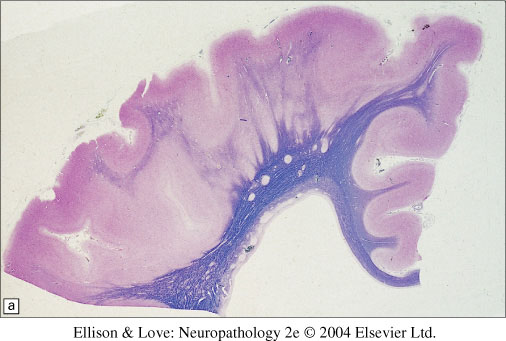
Histopathology
- classically a 4 layered cortex
- outermost molecular layer containing Cajal-Retzius neurons
- Layer 2 of pyramidal neurons with some disorganization
- Layer 3 of lower density of neurons with myelinated axons
- Layer 4 is a broad band of disorganized neurons of variable thickness
- white matter is thinned, with individual or collections of neurons forming heterotopia
- regions can be variable, with normal 6 layered cortex, classic 4 layered cortex, or no clear layering in a disorganized cortex
- other findings include:
- simplification and heterotopia of the inferior olivary nucleus
- cerebellar cortical dysplasia
- corticospinal anomalies
- pachygyria is similar, but also with loss of the grey-white junction (contrast with clear grey-white junction in polymicrogyria)
Experimental models and pathogenesis
LIS1 gene was found to encode a beta-subunit of platelet-activating factor acetylydrolase (PAFAH1B1). Mouse models showed that mutations caused decreased neuronal migration in vitro. Yeast homolog studies showed that this gene product regulates dynein activity, required in nuclear translocation. It is hypothesized that this dysregulation retards cell migration (and not axonal guidance). This dynein activity may also be important in mitotic cell division and cytokinesis.
This could explain the reversal of the cortical layers. Early migration is easier due to a smaller brain, and later migrating neurons end up on the ventricular side because of slowed migration.
RELN mutations also studied via the Reeler mouse. Reelin is an extracellular matrix molecule, and mouse models have implicated it in signaling for cell migration.
Type II Lissencephaly (Cobblestone)
- Type II lissencephaly is entirely genetically, embryologically, and pathologically distinct from type I. Cortical surface has a cobblestone appearance.
- Many type II lissencephalies have associated cerebellar and ocular abnormalities and congenital muscular dystrophy.
- Five syndromes currently described:
- Walker-Warburg Syndrome (WWS) - also called cerebro-ocular dysplasia-muscular dystrophy, Warburg syndrome, hydrocephalus, agyria, retinal dysplasia, and encephalocele syndrome (HARD+/-E), Chemke syndrome, Cerebro-oculo myscular syndrome, Walker type, cerebro-ocular dysgenesis
- Muscle-Eye-Brain Disease (MEB)
- Fukuyama congenital muscular dystrophy (FCMD)
- Congenital muscular dystrophy type 1D (MDC1D) - 1 patient
- mutation in Fukutin related protein (MDC1C)
- all involve defect in O-mannosylation (glycosylation disorders)
Epidemiology
- FCMD is almost exclusively in Japan
- MEB is mostly in Finland
- WWS is found worldwide
- MDC1C and MDC1D have only few reported cases
- FCMD is most common, with incidence of 3 per 100,000
- M = F
Genetics
- Autosomal recessive inheritance for all known types.
- Fukuyama congenital muscular dystrophy
- Fukutin (chr 9) mutation has 3/100,000 incidence, thus 1/90 Japanese are heterozygous
- 87% of defective genes come from single ancestral founder
- other mutations exist in Japan and otherwise
- mouse models show fukutin is required for development of brain, eye, and muscle
- Muscle-eye-brain disease
- POMGnT1 mutation (O-mannose b-1,2-N-acetylglucosaminyltransferase) (chr 1p32)
- mostly in Finland
- Walker-Warburg syndrome
- POMT1 mutation (protein O-mannosyltransferase 1) is one of the known mutations
- some mutations in Fukutin also
- MDC1C
- Fukutin related protein (FKRP) in congenital muscular dystrophy MDC1C and limb girdle muscular dystrophy LGMD2I.
- MDC1D
- acetylglucosaminyl transferase-like protein (LARGE) mutation found in one patient with congenital muscular dystrophy, profound mental retardation, whilte matter changes, and subtle structural abnormalities on MRI
Clinical features
- combination of muscular dystrophy and CNS involvement
- muscular dystrophy usually presents in 1st year of life
- Walker-Warburg - most severe of the types, presents with hypotonia from birth of proximal and distal muscles, development of contractures, survival < 2 years
- Fukuyama - muscle weakness and hypotonia by 9 months. Poor suck and weak cry from birth, lower extremity contractures by 1 year. Muscle pseudohypertrophy in cheeks, calves, forearms. Unable to ambulate, bedridden by 10 years, die by 20 years
- Muscle-eye-brain - similar but more severe than Fukuyama, but survival longer than Fukuyama. Can live to adulthood.
- mental retardation, seizures, hydrocephalus also common
- 20% of WWS have Dandy-Walker malformation, rest have cerebellar vermal hypoplasia. 5-10% have occipital encephalocele
- Muscle-eye-brain and Walker-Warburg have eye anomalies (congenital myopia, congenital glaucoma, pallor of optic discs, retinal hypoplasia/dysplasia. Fukuyama has milder eye defects.
- MRI shows thickened cerebral cortex interpreted as polymicrogyria or pachygyria, and T2 hyperintensity in the white matter (delayed myelination)
Embryology
- over migration of neurons and glia beyond the glial-pial limitans
- from abnormal glycosylation of proteins like alpha-dystroglycan, leading to failure of formation of the glial-pial limitans, thus allowing extension of the radial glial processes
- Result is migration of immature neurons and glia beyond the preplate
Macroscopy
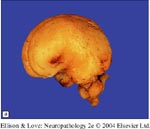
- Only Walker-Warburg, Muscle-Eye-Brain, and Fukuyama syndromes have had pathological reporting
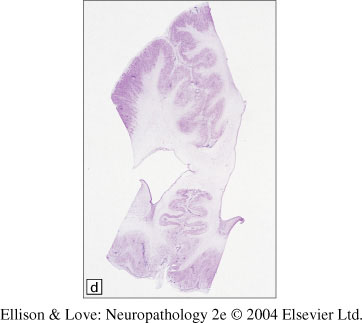
- all have small brains, with loss of sulci and broadening of gyri
- surface of the brain is "lumpy and bumpy" unlike the smooth surface of type I lissencephaly
- WWS may show sparing of mesial temporal cortex and agenesis of the corpus callosum
- FKMD is worse in the lateral posterior temporoparietal and occipital lobes
- Usually there is a gradient between normal and abnormal cortex, and no clear demarcation
- fusion across the interhemispheric fissure or the Sylvian fissure possible
- occipital encephaloceles can occur with occipital lobe herniation
- small cerebellum, especially the vermis, and possibly a Dandy-Walker malformation
- fused cerebellar folia possible, giving matte appearance to cerebellar surface, white matter tracts running over surface of cerebellum
- hydrocephalus
- relative preservation of the deep nuclei
- reduced white matter volume, thickened grey matter
- less affected cortex resembles polymicrogyria
- preserved grey-white junction, but irregular and nodular
- brainstem is small (hypoplasia of corticospinal tracts), normal inferior olives
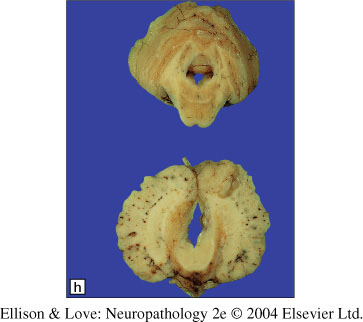
Histopathology
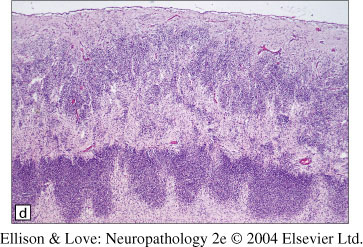
- highly disordered structure, with neurons in the superficial cortex intermingling with white matter tracts
- leptomeninges fused into the cortex, undulating blood vessels
- no defined cortical lamination
- milder areas have fusion of molecular layer like in polymicrogyria
- well defined junctions between normal and abnormal histology
- clear disruption of the pial-glial limitans
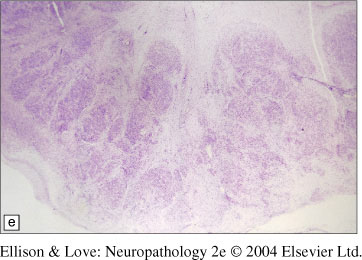
- disorganization of the cerebellum termed "cerebellar polymicrogyria"
- bands of white matter tracts through the surface of the cerebellum
- muscle shows congenital muscular dystrophy - degenerating and regenerating fibers, marked fibrosis
- retinal dysplasias and cataracts
Pathogenesis
- glycosylation involved in 1% of human gene products
- glycosylation is important in enhancing protein stability, mediating ligand-receptor interactions, ensures the fidelity of proteins targeted for secretion
- two types of glycosylation: N-linked glycosylation of asparagine and O-linked glycosylation of serine or threonine
- hypoglycosylation of dystroglycans seen
- dystroglycans important in maintaining muscle sarcolemmal integrity and in brain development
- postulated that type II lissencephalies are due to dysfunction of the glycosylation of dystroglycans