Mitochondrial Encephalomyopathies
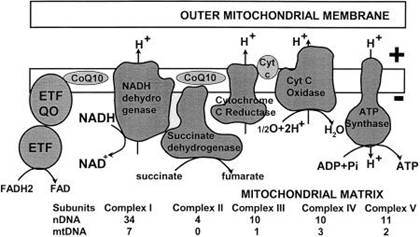
Mitochondrial
functions:
•
cellular energy metabolism
•
cell
signaling
•
apoptosis
•
intermediary
metabolism
•
metabolism
of AA, lipids, cholesterol, steroids and nucleotides
•
intracellular
sequestration of calcium
•
detoxification
of ammonia
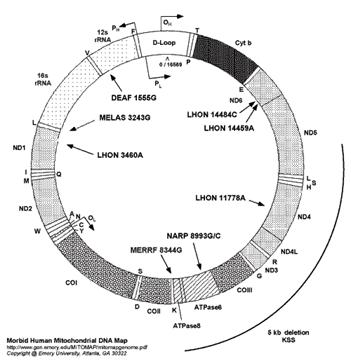
Mitochondrial
genetics:
•
circular
double stranded ring of DNA
•
2
ribosomal RNAs, 22 transfers RNAs,
13 polypeptides of complexes I, III, IV, and V of the respiratory chain
•
2-10 mtDNA rings per mitochondria
•
cells
contain thousands of mitochondria

Clinical
testing:
•
resting
serum lactate – often elevated due to increase anaerobic metabolism
•
CPK –
muscle involvement
•
TSH,
glucose, Hgb A1C – endocrine dysfunction
•
serum
AA, urine organic acids, VLCFA, serum biotinidase –
metabolic screen
•
CSF
lactate – more sensitive than serum
•
ECG –
cardiac involvement common
•
EEG –
may show slowing
•
audiography – often have impaired hearing
•
EMG/NCS
– help to localize problem to muscle
•
CT/MRI
– often show atrophy and related ventriculomegaly,
white matter changes, calcification (esp brain stem
and BG)
•
Muscle
Biopsy
•
H&E, Succinate Dehydrogenase, Modified Gomori-Trichrome,
Cytochrome C Oxidase
•
Molecular
Studies (Southern Blot, PCR, biochemistry on cultured fibroblasts for
respiratory complex activity)

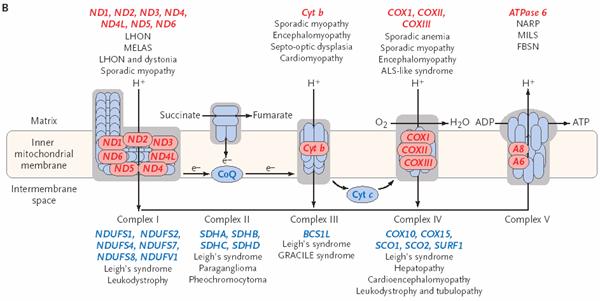
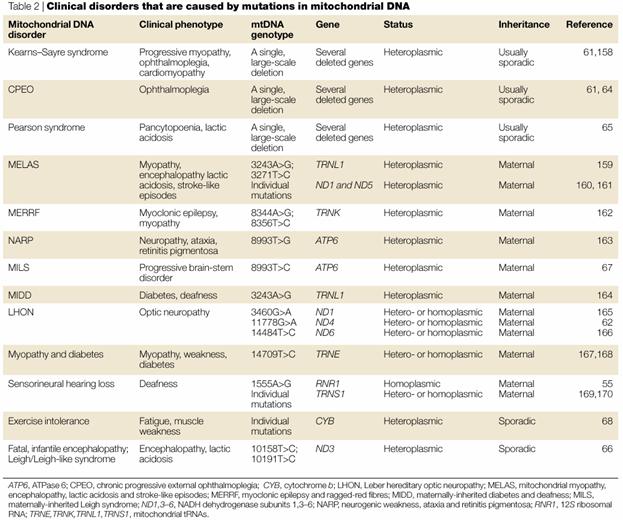
“Common” mitochondrial
encephalomyopathies:
LHON – Leber’s hereditary
optic neuropathy
- Acute or subacute,
painless, central visual loss in 3rd-4th decade
- Also dystonia
and cardiac conduction defects
- Fundoscopy- peripapillary
telangiectasia, microangiopathy,
pseudoedema, tortuous retinal vessels
MELAS – Mitchondrial
encephalopathy with lactic acidosis and stroke-like episodes
- Stroke-like episodes, muscle weakness,
and encephalopathic episodes with high lactate
- Also seizures, dementia, short
stature, migraines, episodic vomiting
MERFF – Myoclonic epilepsy
with ragged red fibers
- Progressive myoclonic
epilepsy, action myoclonus, ataxia
- Progressive muscle weakness/atrophy, hypertrophic cardiomyopathy,
dementia, deafness, multiple lipomata
- Ragged red fibers on muscle biopsy
Leigh syndrome
- Not classically one genetic disorder,
but arise from different mutations
- Developmental
delay/regression, central hypoventilation, early hypotonia/late
spasticity
- Optic atrophy, pigment degeneration, nystagmus, ataxia, dystonia,
multifocal myoclonus, seizures
Kearns-Sayre syndrome
- Caused by large deletions in mtDNA
- Progressive external ophthalmoplegia, pigmentary
retinopathy, complete heart block, cerebellar
ataxia, CSF protein > 100 mg/dl
- Progressive external ophthalmoplegia (PEO) can occur independently
Management of
mitochondrial disorders:
- Symptomatic support for seizures,
endocrine abnormalities, etc.
- Unclear role for coenzyme Q10, biotin,
thiamine, vitamins C & K3, carnitine, dichloroacetate
Reference: Taylor RW & Turnbull DM.
(2005). Mitochondrial DNA mutations in human disease. Nature Rev Gen.
6:389-402.